Болезнь Вильсона-Коновалова
Диагностика
Диагностика гепатолентикулярной дегенерации включает в себя визуальный осмотр, оценку симптомов, пальпаторное обследование печени, а также лабораторные исследования. Во время личного осмотра врач уточняет у пациента, есть ли у него наследственная предрасположенность к гепатолентикулярной дегенерации, осматривает глаза на предмет выявления характерного коричневого кольца. При пальпации отмечается гепатомегалия.
При диагностике гепатолентикулярной дегенерации используются такие лабораторные методики:
- ДНК-маркеры. Данный метод диагностики даёт возможность точно установить наличие этого наследственного недуга;
- биохимия крови. Позволяет определить уровень меди в крови;
- биопсия печени;
- анализ мочи на наличие в ней меди.
Существует ещё один информативный метод диагностики патологии у детей и взрослых – использование меченой меди. Её вводят в организм, а потом наблюдают, в каком органе она будет накапливаться.
Возможности лечения
Приведенные сроки продолжительности жизни пациентов при современном уровне медицины удается значительно увеличить при помощи раннего начала лечения болезни Вильсона-Коновалова и использования всех возможных средств.
Питание пациента должно проводиться согласно диетическому столу №5а (для заболеваний печени). Исключаются все острые блюда, жареные и жирные. Одновременно для ограничения поступления меди с пищей категорически запрещаются: изделия и натуральные продукты, содержащие шоколад, орехи, кофе, мясо печени, раков и кальмаров, сухофрукты, бобовые, крупы из цельной пшеницы.
Основное направление — обеспечение вывода излишков меди. Главным препаратом считается D-пеницилламин. Препарат назначается с небольшой дозы по схеме, затем она увеличивается. Необходимо учитывать пожизненный прием лекарства и его негативные свойства.
Специалисты обращают внимание на низкое качество отечественного Пеницилламина, его токсичность. У некоторых пациентов с гепатолентикулярной дегенерацией побочные явления заключаются в дерматитах, анемии
Другое направление — терапия солями цинка (сульфат, оксид). Применение комбинаций D-пеницилламина с препаратами цинка позволяет проводить терапию низкими дозами и избегать отрицательных последствий. Если заболевание выявлено до начала клинических проявлений, то рекомендуются только препараты цинка. Дополнением является использование Унитиола.
В обязательном порядке пациенту назначаются гепатопротекторные средства: витамины, Эссенциале. Для поддержки проводимости в нервных волокнах необходимы витамины группы В. Предложен способ био-гемоперфузии крови с живыми изолированными клетками печени и селезенки. Методику называют «вспомогательной печенью».
Она проводится в специализированных центрах при безуспешной терапии для поддержки организма пациента до операции трансплантации печени. Пересадка здоровой печени донора помогает решить множество проблем лечения.
Сама операция требует подготовки больного, учета рисков в связи со сниженной свертываемостью крови. Это наиболее радикальный метод лечения форм болезни с преобладающим поражением печени.
Эффективность лечения значительно улучшается при ранней диагностике. Удается достичь уменьшения проявлений. Пациенты остаются социально адаптированными людьми: способны полностью к самообслуживанию, учатся, работают по профессии, создают семью. Имеются наблюдения за молодыми женщинами с болезнью Вильсона-Коновалова, выносившими беременность и родившими здоровых детей.
Раз эффект лечения болезни зависит от раннего выявления и начала, то семьям, в которых есть ребенок с гепатолентикулярной дегенерацией, необходимо обследовать его братьев и сестер с применением современных молекулярно-генетических методик. Знание будущих родителей о своем носительстве предотвратит рождение больного малыша.
Лечение этого недуга является трудной задачей даже для опытных специалистов. Болезнь Вильсона-Коновалова — это синдром, объединяющий поражение головного мозга и других органов и систем вследствие накопления в организме меди. Патологию называют также гепатолентикулярной дегенерацией, поскольку первой на ее возникновение реагирует печень. Существует и другое синонимическое название болезни — гепатоцеребральная дистрофия. Она указывает на поражение гепатоцитов и центральной нервной системы. Лечение болезни длительное и трудное, а прогноз относительно выздоровления пациента неблагоприятный.

Консервативное лечение
При обнаружении патологии на ранней стадии возможно немедикаментозное лечение. Здесь может помочь соблюдение диеты, которую необходимо организовать так, чтобы в организм поступало минимальное количество меди вместе с пищей. Запрещается пить обычную воду, а необходимо употреблять только дистиллированную.
После диагностики заболевания обязательно назначается лекарственная терапия, которая помогает сдерживать симптомы гепатолентикулярной дегенерации. Из препаратов используются:
- Хелаты, которые помогаю выводить медь из организма.
- Препараты, которые блокируют всасывание меди из желудка.
- Иммуносупрессоры.
- Противовоспалительные средства.
- Поливитамины и микроэлементы.
- Препараты цинка.
- При нарушении неврологических функций – препараты, помогающие контролировать их развитие.
- Гепатопротекторы.
- Антиоксиданты.
Назначенные дозировки нельзя изменять самостоятельно, а также нельзя по своему желанию отменять то или иное лекарственное средство.
По жизненным показаниям может быть проведено хирургическое вмешательство, и чаще всего здесь применяется пересадка печени.
Генетика
Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот
Ген болезни Вильсона — Коновалова(ATP7B) расположен в длинном плече 13-й хромосомы (13q14.3). Ген кодирует P-тип АТФазы, которая транспортирует медь в жёлчь и включает её в церулоплазмин. В 10 % случаев мутация не обнаруживается.
Хотя описано почти 300 мутаций ATP7B, в большинстве популяций болезнь Вильсона возникает в результате небольшого количества мутаций, специфичных для этих популяций. Например, для западных популяций мутация H1069Q (замена гистидина на глутамин в позиции 1069 белка) присутствует в 37-63 % случаев заболевания, в то время как в Китае эта мутация очень редка и R778L (замена аргинина на лейцин в позиции 778) встречается чаще. Относительно мало известно о влиянии мутаций на течение заболевания, хотя по данным некоторых исследований мутация H1069Q предполагает более позднее начало неврологических симптомов.
Нормальные вариации в гене PRNP могут изменить течение болезни, увеличивая возраст появления заболевания и влияя на тип симптомов, которые развиваются. Этот ген кодирует прионный белок, который активен в головном мозге и других тканях, а также, как полагают, участвует в транспорте меди.
У заболевания аутосомно-рецессивный тип наследования. То есть больной должен получить дефектный ген от обоих родителей (см. на рисунке). Люди только с одним мутантным геном называются носителями (гетерозиготы). У них могут возникать слабовыраженные нарушения метаболизма меди.
Медицинские новости
Pfizer рассказывает о транстиретиновой амилоидной кардиомиопатии
14.11.2019
Специалисты сходятся во мнении, что необходимо привлечение внимания общественности к проблемам сердечно-сосудистых заболеваний. Некоторые из них являются редкими, прогрессирующими и трудно диагностируемыми. К таким относится, например, транстиретиновая амилоидная кардиомиопатия
В России проходят дни МНО
14.10.2019
12, 13 и 14 октября, в России проходит масштабная социальная акция по бесплатной проверке свертываемости крови – «День МНО». Акция приурочена к Всемирному дню борьбы с тромбозами.
Заболеваемость менингитом в России растет
07.05.2019
Заболеваемость менингококковой инфекцией в РФ за 2018 г. (в сравнении с 2017 г.) выросла на 10 % (1). Один из распространенных способов профилактики инфекционных заболеваний – вакцинация. Современные конъюгированные вакцины направлены на предупреждение возникновения менингококковой инфекции и менингококкового менингита у детей (даже самого раннего возраста), подростков и взрослых.
Что делать при укусе клеща, или как этого избежать
25.04.2019
Грядут долгие выходные, и, многие россияне поедут отдыхать за город. Не лишним будет знать, как защитить себя от укусов клещей. Температурный режим в мае способствует активизации опасных насекомых…
Как защитить себя и своих близких от коклюша?
05.04.2019
Заболеваемость коклюшем в РФ за 2018 г. (в сравнении с 2017 г.) выросла почти в 2 раза 1, в том числе у детей в возрасте до 14 лет. Общее число зарегистрированных случаев коклюша за январь-декабрь выросло с 5 415 случаев в 2017 г. до 10 421 случаев за аналогичный период в 2018 г. Заболеваемость коклюшем неуклонно растет с 2008 года…
Каким образом наследуется болезнь
Тип наследования аномального гена болезни называется аутосомно-рецессивным. Это означает, что заболеть могут только дети, получившие сразу два мутантных гена (от матери и отца). Родители являются гомозиготными носителями.
Если ребенок получает ген только от одного из родителей, то он не заболевает, но становится гетерозиготным носителем. У него возможны отклонения в обмене меди, но слабо выраженные. Насколько здорово будет третье поколение, зависит от встречи с аналогично измененной структурой хромосом жены или мужа.
Известен ген болезни — АТР7В — он обеспечивает синтез транспортирующего медь белка (церуллоплазмина). Находится на длинном плече в хромосоме №13, на участке с кодом 13q14-q21.
Обнаружены разные виды мутаций у пациентов в Китае и странах Запада. Причины нарушений остаются неясными. У 10% больных вообще не выявлены генные изменения
Установлено, что в проявлении заболевания важное значение принадлежит факторам, поражающим печень. К ним относятся перенесенные инфекционные болезни (вирусный гепатит), интоксикация
Роль меди в норме и при гепатолентикулярной дегенерации
Здоровый человек получает ежедневно с пищей 2 мг меди, из них усваивается только 1/3 часть, но и этого вполне хватает для обеспечения потребности организма.
Медь необходима печени:
- для синтеза белков и ферментов, а значит для роста, физического и умственного развития;
- образования гемоглобина, транспорта железа;
- синтеза эритроцитов и лейкоцитов;
- обеспечения эластичности стенок сосудов за счет участия в синтезе коллагена;
- стимуляции гормональной активности гипофиза и инсулина;
- выработки клеток иммунитета;
- образования антиоксидантов, предотвращения раннего старения.
Попадая их тонкого кишечника в кровь, по воротной вене молекулы меди доставляются в печеночные клетки. Здесь часть связывается с белком церулоплазмином и возвращается в кровяное русло, другая (лишняя) — переходит в желчь и удаляется из организма. Одна молекула церуллоплазмина включает 8 атомов меди.
В патогенезе болезни Вильсона-Коновалова нарушаются обе функции. Церулоплазмин разрушается. Кровь больного переполняется значительным количеством несвязанной меди. Она откладывается в разных органах: в печени, головном мозге, почках, роговице глаз, переходит в мочу.
В гепатоцитах печени, как ответная реакция на внедрение меди образуется воспаление, переходящее в узловой цирроз. В почечной ткани страдают проксимальные канальцы. Повреждение головного мозга выражается в поражении базальных ганглиев, ядер мозжечка и черной субстанции.
В тканях глаза медь откладывается вокруг роговой оболочки и образует видимое кольцо Кайзера-Флейшера. Преимущественное отложение меди в одном из органов зависит от возраста. Для детей типично поражение печени. После 20 лет основные разрушения локализуются в головном мозге.
Патологическая анатомия
В головном мозге при гепато-церебральной дистрофии размягчается чечевицеобразное ядро, особенно скорлупа, с образованием мелких кист. Поражаются и другие образования: хвостатое ядро, глубокие слои коры, мозжечок, в частности зубчатые ядра, подбугорные ядра; в остальных отделах головного мозга изменения выражены меньше.
Все изменения делятся на ангиотоксические и цитотоксические. Первые выражаются в атонии сосудов, особенно мелких, и изменении их стенок. В результате возникают стазы, распространённый периваскулярный отек с аноксией нервной ткани и её гибелью; часты геморрагии и следы их в виде скоплений гемосидерина.
Цитотоксический компонент заключается в распространённых дистрофических изменениях макроглии нервных клеток, часто заканчивающихся их гибелью. Характерно появление глии Альцгеймера, которая образуется из обычных астроцитов. Нередко встречаются изменённые нервные клетки, очень похожие на глию Альцгеймера; сходные клетки обнаруживаются также в печени и почках. В основе этих клеточных изменений лежит один и тот же фактор — однотипное нарушение клеточного обмена, вероятно, обмена нуклеиновых кислот.
Чем позднее начинается заболевание, тем медленнее оно протекает, тем более диффузны изменения в головном мозге и тем более цитотоксический компонент преобладает над ангиотоксическим.
Печень вследствие атрофического цирроза уменьшена и бугристая; участки нормальной ткани чередуются с участками некротическими, дегенерирующими и с островками регенерации; обильное новообразование сосудов приводит к появлению анастомозов между ветвями воротной и нижней полой вены.
История править править код
Английский невролог Сэмюель Вильсон (англ. S. Wilson — более нормативная передача У́илсон) (1878 — 1937) в 1912 году описал типичные для гепато-церебральной дистонии изменения в головном мозге, установил постоянное наличие цирроза печени и дал описание клиники нового заболевания, названного им прогрессивной лентикулярной дегенерацией (лат. lenticularis чечевицеобразный).
В качестве основных симптомов заболевания были отмечены разнообразные непроизвольные движения в конечностях и туловище, мышечная ригидность, приводящая к скованности, дисфагия и дизартрия, аффектные вспышки, иногда психические расстройства, но признаки поражения пирамидных путей отсутствовали. Ещё раньше К. Вестфалем (1883) и А. Штрюмпелем (1898) было описано заболевание, которое по клиническому сходству с рассеянным склерозом получило название «псевдосклероз». Заболевание характеризовалось распространёнными, размашистыми, ритмичными непроизвольными движениями, повышением мышечного тонуса, амимией, дизартрией и выраженными психическими нарушениями вплоть до такого расстройства интеллекта, как слабоумие.
В дальнейшем оказалось, что прогрессивная лентикулярная дегенерация и псевдосклероз являются разными формами одного и того же заболевания, которое Галль (1921) назвал гепато-лентикулярной дегенерацией. Однако изменения в мозге при нём никогда не ограничиваются лентикулярными ядрами и нередко бывают даже сильнее выражены в других отделах мозга.
В 1960 году советский невропатолог Н. В. Коновалов предложил название «гепато-церебральная дистрофия», значительно расширил представления о патофизиологии, патогенезе и клинике этой болезни и выделил 4 формы поражения нервной системы и одну абдоминальную.
Формы и симптомы болезни
В зависимости от основных признаков выделяют три основные формы болезни Вильсона — Коновалова. Это патология, приводящая к серьезным поражениями печени; заболевание, поражающее нервную систему; смешанная форма. В соответствии с этими типами у пациента преобладают те или иные симптомы.
Печеночная форма
Печеночная или брюшная форма болезни Вильсона развивается у людей до 40 лет и характеризуется поражением печени, схожим с циррозом. Кроме того, пациенту диагностируют хронический гепатит. В 80% случаев эта форма имеет следующие симптомы:
- метеоризм;
- снижение уровня активности;
- тупая боль в правом подреберье;
- увеличение количества жидкости в брюшной полости;
- периодические носовые кровотечения;
- утолщение пальцев ног и рук;
- желтуха;
- лихорадка;
- увеличение размеров селезенки.

Неврологическая форма
Эта форма болезни отличается проявлениями в очень раннем возрасте: мышечной ригидностью, нарушениями речи, небольшим постепенным снижением интеллектуальных способностей. Наблюдаются периоды обострения и ремиссии. У пациентов в возрасте 10-25 лет отмечают тремор, брадилалию. Они медленно пишут, читают, разговаривают, бесцельно повторяют движения руками.
Редкие симптомы
У 15% пациентов отмечают такие главные симптомы, присутствующие как комплексно, так и по отдельности:
- гемолитическая анемия;
- поражение почек;
- посинение или гиперпигментация кожных покровов и ногтей;
- хрупкость костей, приводящая к постоянным переломам;
- артроз;
- глухота;
- гинекомастия.

Течение болезни Вильсона
Существуют острый и хронический клинические типы течения заболевания. Кроме того, врачи определяют латентную стадию, которая длится не более 7 лет. При этом все основные симптомы присутствуют, но не сказываются на качестве жизни, так как выражены слабо. Иногда заболевание практически не проявляет себя до 5 лет. Пик болезни приходится на 8-15 лет, но уже с рождения диагностируют проблемы с печенью. Развитие патологии имеет свои особенности в зависимости от типа течения:
- Острое. Недуг доставляет немало беспокойств уже в раннем возрасте, все симптомы обострены и очень быстро приумножаются. Организм человека угасает очень быстро, лечение почти не помогает и не облегчает состояние. Быстрый летальный исход неизбежен в 90% случаев.
- Хроническое. Болезнь развивается медленно, сначала может быть латентной. Поражается сначала печень, после — органы нервной системы. В юношеском возрасте у пациентов отмечают нарушение походки и координации движений, редко — истерию. Выраженность всех симптомов средняя, прогрессируют они медленно.
Клиническая картина и течение
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет хроническое прогрессирующее течение. Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств (желтуха, боли в правом подреберье, диспептические явления). Порой развивается выраженный гепато-лиенальный синдром. Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов и расстройств психики. Пирамидные симптомы могут быть, но чаще отсутствуют. Чувствительность обычно не нарушена.
Типичным симптомом болезни являются кольца Кайзера-Флейшера — отложения по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента, более выраженные на поздних стадиях. Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления (кровоточивость дёсен, носовые кровотечения, положительная проба жгута), мраморность кожи, акроцианоз. Капилляроскопия обнаруживает атонию капилляров и застойность кровотока. Отмечаются суставные боли, профузные поты, остеопороз, ломкость костей.
Патология печени клинически выявляется примерно у 30 % больных, а в ряде случаев она может быть обнаружена только функциональными пробами, например пробой с нагрузкой галактозой, пробой Квинка, пробой Бергмана-Эльботта, бромсульфофталеиновой пробой; количество билирубина в крови и уробилина в моче обычно увеличено; изменены осадочные реакции Таката-Ара и Грея, обычны лейкопения, тромбоцитопения, гипохромная анемия.
Различают 5 форм гепато-церебральной дистрофии:[уточнить]
- Брюшная форма — тяжёлое поражение печени, приводящее к смерти раньше появления симптомов со стороны нервной системы; заболевают дети. Её продолжительность от нескольких месяцев до 3-5 лет.
- Ригидно-аритмогиперкинетическая, или ранняя форма — отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
- Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возрасте, течёт медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжёлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
- Дрожательная форма начинается в возрасте 20-30 лет, течёт довольно медленно(10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжёлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
- Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжёлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Наибольшая летальность (50 %) отмечается при печёночной форме с массивным некрозом и гемолизом у детей до 6 лет. Смерть больных от неврологических нарушений при отсутствии лечения наступает через 5-14 лет. Основная причина при этом интеркуррентные заболевания или желудочно-кишечные кровотечения, портальная гипертензия.
Симптоматика
Симптомы гепатоцеребральной дистрофии начинают проявляться ещё у маленьких детей, но пика своей активности и выраженности достигают в возрасте 12–16 лет. Болезнь Вильсона-Коновалова имеет несколько форм развития, которые различаются в зависимости от того, в каком органе наблюдается наибольшее скопление меди.
Печёночная форма гепатоцеребральной дистрофии
Встречается чаще всего – в 80% случаев. Патология протекает по типу хронического гепатита. В результате её прогрессирования развивается цирроз печени и происходит дистрофия органа. Первый симптом, который указывает на возникновение патологии – желтуха. Её появление связано с тем, что в кровь выделяется большое количество билирубина. Сначала начинают желтеть склеры, немного позже – кожный покров. Но кожного зуда при этом не наблюдается. Моча темнеет, а кал обесцвечивается.
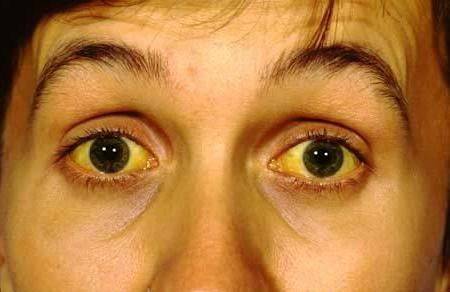 Симптомы болезни Вильсона-Коновалова
Симптомы болезни Вильсона-Коновалова
Если патология продолжает прогрессировать, то развивается асцит и появляются отеки. Размеры живота могут увеличиваться очень быстро, и начинает давить на диафрагму, затрудняя дыхание. Отеки отмечаются на стопах и голенях.
Кровотечения также являются частым симптомом болезни Вильсона-Коновалова. Сначала кровоточат десны, но при нарушении функционирования печени отмечаются кровотечения из пищевода, кишечника.
Поражение ЦНС при болезни Вильсона-Коновалова
Как правило, симптомы при данной форме патологии сочетаются с печёночными проявлениями. Изначально пациенту становится сложно выполнять мелкие движения руками, так как у него развивается тремор. Речь становится стёртой
Если обратить внимание на лицо больного синдромом Вильсона-Коновалова, то можно отметить мелкие непроизвольные подёргивания различных групп мышц. Человек может гримасничать или совершать однотипные движения
На поздних этапах прогрессирования недуга отмечается гипертонус разгибателей в конечностях.
Изменения психологического статуса при гепатолентикулярной дегенерации наблюдаются практически у всех больных. У них возникает бессонница, они замкнуты, ведут себя неадекватно, страдают бредом и галлюцинациями. Также отмечается характерный симптом болезни Вильсона – по наружной окружности радужки глаза отмечается коричневое кольцо.
Редкие формы гепатоцеребральной дистрофии
О многих пациентов наблюдается остеопороз. Данное состояние развивается из-за того, что обмен веществ в организме существенно нарушается, и поэтому вымываются соли кальция. Поэтому у людей с диагнозом гепатолентикулярная дегенерация часто случаются неожиданные переломы.
Из-за отложения большого количества меди в почечных клубочках может нарушаться фильтрация крови. Поэтому при гепатоцеребральной дистрофии в моче появляется сахар, а также эритроциты (моча имеет лёгкий красноватый оттенок).
Синдром Вильсона-Коновалова может протекать на протяжении 7 лет без единого симптома, поэтому его можно сразу и не обнаружить у детей. Но по истечении латентного периода проявляются характерные симптомы недуга. Гепатоцеребральная дистрофия требует проведения специальной терапии.