Болезнь Шарко-Мари-Тута болезнь Шарко Мари Тута, наследственная моторно-сенсорная невропатия I типа, наследственная нейропатия Шарко-Мари-Тута, ШМТ, невральная амиотрофия
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.



- Аденозинтрифосфат натрия (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- Пентоксифиллин (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- Мильгамма (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- Метандростенолон (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- Церебролизин (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
- Галантамин (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Формы болезни Шарко-Мари-Тута
Существует много форм болезни Шарко-Мари-Тута, включая CMT1, CMT2, CMT3, CMT4 и CMTX. CMT1, вызванный аномалиями в оболочке миелина, имеет три основных типа. CMT1A является аутосомно-доминантным заболеванием, которое возникает в результате дублирования гена на хромосоме 17, которая несет инструкции по производству периферического белка миелина-22 (PMP-22). Белок PMP-22 является критическим компонентом миелиновой оболочки. Сверхэкспрессия этого гена вызывает ненормальность структуры и функции миелиновой оболочки. Пациенты испытывают слабость и атрофию мышц нижних конечностей, начиная с подросткового возраста; позже они испытывают слабость рук и сенсорную потерю. Интересно, что другая невропатия, отличная от CMT1A, называемая наследственной невропатией с предрасположенностью к параличу давления (HNPP), вызвана удалением одного из генов PMP-22. В этом случае аномально низкие уровни гена PMP-22 приводят к эпизодической рецидивирующей демиелинизирующей нейропатии. CMT1B является аутосомно-доминантным заболеванием, вызванным мутациями в гене, который несет инструкции по изготовлению нуклеотида миелина (P0), который является еще одним критическим компонентом миелиновой оболочки. Большинство из этих мутаций являются точечными мутациями, что означает, что ошибка встречается только в одной букве генетического кода ДНК. На сегодняшний день ученые выявили более 120 различных точечных мутаций в гене P0. В результате аномалий в P0 CMT1B вызывает симптомы, сходные с симптомами, обнаруженными в CMT1A. Менее распространенные CMT1C, CMT1D и CMT1E, которые также имеют симптомы, сходные с симптомами, обнаруженными в CMT1A, вызваны мутациями в генах LITAF, EGR2 и NEFL соответственно.
Болезнь Шарко-Мари-Тута является результатом аномалий в аксоне периферической нервной клетки, а не в оболочке миелина. Он менее распространен, чем CMT1. CMT2A, наиболее распространенная аксоновская форма CMT, вызвана мутациями в Mitofusin 2, белке, связанном с митохондриальным слиянием. CMT2A также был связан с мутациями в гене, который кодирует белок 1B-бета-члена семейства кинезина, но в других случаях он не реплицируется. Кинезины — это белки, которые действуют как двигатели, чтобы помочь обеспечить транспортировку материалов вдоль клетки. Другие менее распространенные формы CMT2 были недавно идентифицированы и связаны с различными генами: CMT2B (связанный с RAB7), CMT2D (GARS). CMT2E (NEFL), CMT2H (HSP27) и CMT2l (HSP22).
CMT3 или Dejerine-Sottas — тяжелая демиелинизирующая невропатия, которая начинается в младенчестве. У младенцев происходит тяжелая атрофия мышц, слабость и сенсорные проблемы. Это редкое расстройство может быть вызвано определенной точечной мутацией в гене P0 или точечной мутацией в гене PMP-22.
CMT4 включает несколько различных подтипов аутосомно-рецессивного демиелинизирующего двигателя и сенсорных невропатий. Каждый подтип невропатии вызван другой генетической мутацией, может влиять на конкретную этническую популяцию и производит различные физиологические или клинические характеристики. Лица с CMT4 обычно развивают симптомы слабости ног в детстве, а в подростковом возрасте они не могут ходить. Несколько генов были идентифицированы как вызывающие CMT4, включая GDAP1 (CMT4A), MTMR13 (CMT4B1), MTMR2 (CMT4B2), SH3TC2 (CMT4C), NDG1 (CMT4D), EGR2 (CMT4E), PRX (CMT4F), FDG4 (CMT4H) и фиг.4 (CMT4J).
CMTX возникает точечной мутацией гена connexin-32 на Х-хромосоме. Коннексин-32-белок экспрессируется в клетках Schwann-клеток, которые обертывают нервные аксоны, составляя один сегмент миелиновой оболочки. Этот белок может участвовать в связи с клеткой Шванна с аксоном. Мужчины, которые наследуют один мутированный ген от своих матерей, проявляют умеренные или тяжелые симптомы заболевания, начиная с позднего детства или подросткового возраста (Y-хромосома, которую самцы наследуют от своих отцов, не имеет ген коннексин-32). Женщины, которые наследуют один мутированный ген от одного родителя и одного нормального гена от другого родителя, могут проявлять мягкие симптомы в подростковом возрасте или позже или вообще не могут развиваться симптомы заболевания.
Возможные осложнения ШМТ
Дыхание может быть затруднено, если болезнь поражает нервы, контролирующую диафрагму. Пациенту может понадобиться бронхолитические лекарственные средства или искусственная вентиляция легких. Избыточный вес или ожирение может затруднять дыхание.
Депрессия может быть результатом психического стресса, тревоги и разочарования жизни с любым прогрессирующим заболеванием. Когнитивная поведенческая терапия помогает пациентам лучше справляться с повседневной жизнью и, при необходимости, с депрессией.
Хотя ШМТ нельзя вылечить, некоторые меры помогут избежать дальнейших проблем. Они включают в себя хороший уход за ногами, так как существует повышенный риск травмы и инфекции, отказ от кофе, алкоголя и курения.
Понравилась новость? Читайте нас в Facebook
Приглашаем подписаться на наш канал в Яндекс Дзен
Как диагностировать ШМТ
Врач спросит о семейном анамнезе, и выявит признаки мышечной слабости — снижение мышечного тонуса, плоскостопие или высокий свод стоп (кавус).
Для исследования нервной проводимости проводится измерение силы и скорости электрических сигналов, которые проходят через нервы (Электромиография). Электроды помещаются на кожу, и вызывают слабые поражения электрическим током, которые стимулируют нервы. Задержанный или слабый ответ предполагает расстройство нервной системы, и, возможно, ШМТ.
При электромиографии (ЭМГ) тонкую иглу вводят в мышцы. Когда пациент расслабляет или сокращает мышцы, измеряется электрическая активность. Тестирование различных мышц покажет, какая из них страдает.
Генетическое тестирование проводится с помощью пробы крови, которая может показать, имеет ли пациент мутации гена.
Симптомы
Клиническая картина болезни имеет общие симптомы независимо от типа, но при этом может проявляться индивидуально. Даже в одной семье, когда заболевание провоцируется одним и тем же геном, у двух близких родственников оно далеко не всегда проявляется одинаково.
Общие симптомы болезни Шарко:
- атрофия мышц дистальных отделов конечностей (голень, предплечье);
- появление нечувствительных участков, онемение (без «мурашек» и покалываний);
- нарушение рефлексов;
- увеличение свода стопы (утолщение голеностопного сустава);
- сколиоз, кифоз, другие деформации скелета.
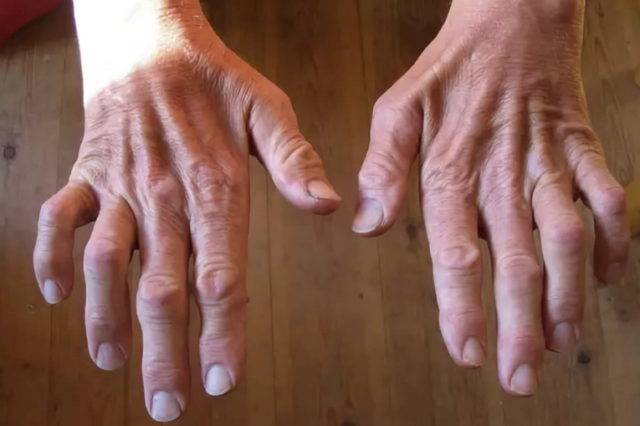
Дополнительные симптомы при 1-м типе болезни:
- сильные судороги в мышцах голени (чаще в передних отделах) после физической нагрузки;
- нарастающая слабость мышц;
- изменение походки, у детей – ходьба на цыпочках;
- нарушение тонуса мышц-сгибателей и разгибателей;
- молоткообразная форма пальцев ног (вместе с увеличением свода стопы);
- атрофия мышц ног, начиная с дистальных (дальних) отделов – от стопы к колену; «аистообразная», «бутылкообразная» форма ног;
- по мере развития болезни – дрожание рук, слабость пальцев, нарушение мелкой моторики;
- нарушение сухожильных и периостальных рефлексов в области кистей и стоп;
- нарушение вибрационной, тактильной, а затем термальной и болевой чувствительности кистей и стоп;
- утолщение нервных стволов;
- искривления позвоночника;
- возможна атипичная форма болезни в виде синдрома Руси-Леви: топтание при попытке стоять неподвижно, неустойчивость при ходьбе, сильное дрожание рук в статичном положении, ночные судороги в икроножных мышцах, парезы.
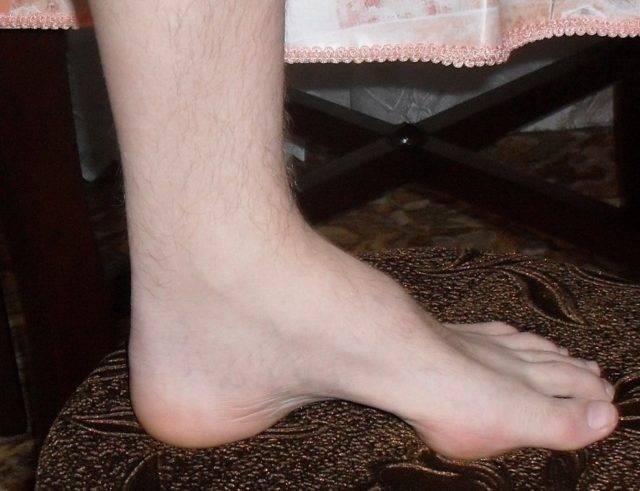
Особенности 2-го типа заболевания:
- изменения подъема стопы и формы пальцев встречаются реже, чем при 1-м типе;
- не наблюдаются утолщения нервных стволов;
- меньшая степень нарушения чувствительности;
- наблюдается синдром беспокойных ног перед сном;
- реже встречается ослабевание мышц в руках;
- возможно нарушение слуха (когда болезнь передается по женской линии);
- транзисторная энцефалопатия после физической активности на высоте: нарушения речи, шатание при ходьбе, затрудненное глотание, слабость проксимальных (ближних к туловищу) отделах конечностей.
Проявлением данной патологии обоих типов могут стать аутоиммунные реакции, когда вырабатываемые организмом специальные антитела разрушают миелиновые оболочки нервных волокон.
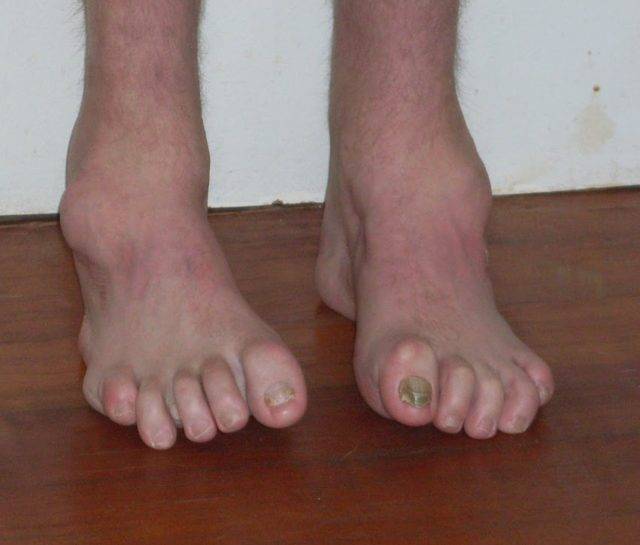
Симптомы болезни Шарко-Мари-Тута
Дебют заболевания отмечается в возрасте 10–20 лет. Первоначально появляются слабость в дистальных отделах ног, утомляемость в мышцах ног при длительном стоянии (постепенно нарастают на протяжении десятков лет). Позже может присоединяться боль в мышцах голени после долгой ходьбы (70%). При ходьбе приходится высоко поднимать ноги. Онемение в стопах отмечается в 80% случаев. Мышечная слабость в руках появляется спустя 10–15 лет после начала заболевания.
При объективном осмотре выявляют симметричную мышечную слабость в перонеальной группе (свисающая стопа) (до 100%), в мышцах кистей рук (40%). Определяются симметричные мышечные атрофии дистальных отделов ног («ноги аиста») (рис. 1), реже в кистях рук («когтистая кисть»). Отмечают угнетение ахилловых рефлексов, позже исчезают коленные рефлексы, после — карпорадиальные; нарушение чувствительности в кистях/стопах («высокие носки», «перчатки») (80%); изменение походки («степпаж», ходьба на пятках невозможна); сколиоз/кифосколиоз, поясничный гиперлордоз, высокий свод стопы (pes cavus) (50%) (рис. 2).
| Рис. 1 | Рис. 2 |
Диагностика
Диагностическая процедура включает ряд методов, среди которых:
- подробный личный и семейный анамнез;
- клиническая оценка мышечной силы, чувствительности;
- электрофизиологическое исследование скорости проводимости нервного волокна;
- неврологическое исследование.
Наиболее распространенные формы заболевания могут быть диагностированы путем анализа ДНК из крови пациента.
При диагностике необходимо тесное сотрудничество невролога, генетика, реабилитолога, хирурга-ортопеда и протезиста. В соответствии с выводами обследования, даются рекомендации относительно индивидуального плана реабилитации, при необходимости, назначается ортопедическая операция.
Значительная изменчивость клинических признаков болезни, наряду с отсутствием знаний о ней у многих врачей, часто приводит к неправильной диагностике.
Симптомы ШМТ у взрослых людей
- Слабость в мышцах ног и лодыжек;
- Искривление пальцев ног;
- Трудности подъема стопы из-за слабых мышц голеностопного сустава;
- Онемение в руках и ногах;
- Изменение формы голени, при этом нога становится очень тонкой ниже колена, в то время как бедра сохраняют нормальный объем мышц и форму (нога аиста);
- Со временем руки ослабевают и пациентам трудно выполнять повседневную работу;
- Появляются боли в мышцах и в суставах, человеку тяжело ходить. Нейропатическая боль возникает вследствие поврежденных нервов;
- В тяжелых случаях пациент может нуждаться в коляске, в то время как другие могут использовать специальная обувь или другие ортопедические устройства.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При ШMT 1 мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга. Это приводит к мышечной слабости и потере чувствительности или онемению.
При ШМТ 2 мутирующий ген влияет непосредственно на аксоны. Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
ШМТ 3 или болезнь Дежерин-Соттас, редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4 является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.